
1 Introdução
Os físicos chamam a função que representa um elétron confinado em um potencial central de função de onda. Os químicos preferem chamar essa mesma função de orbital.
É importante conhecer as duas linguagens: se expressar através das funções de onda e por meio dos orbitais. Por exemplo, a soma das funções de onda \(\frac{1}{\sqrt{2}}(\psi_{21-1} - \psi_{211})\), sendo:
\[ \psi_{21-1} = \frac{1}{\sqrt{64\pi}} \left( \frac{Z}{a} \right)^{\frac{5}{2}} r e^{-Zr/2a} \sin{\theta}e^{-i\phi} , \tag{1.1} \]
\[ \psi_{211} = -\frac{1}{\sqrt{64\pi}} \left( \frac{Z}{a} \right)^{\frac{5}{2}} r e^{-Zr/2a} \sin{\theta} e^{i\phi} , \tag{1.2} \]
é o orbital \({\rm 2p_x}\):
\[ {\rm 2p_x} = \frac{1}{\sqrt{32\pi}} \left( \frac{Z}{a} \right)^{\frac{5}{2}} r e^{-Zr/2a} \sin{\theta} \cos{\phi} . \tag{1.3} \]
Já o orbital \({\rm 2p_y}\) é a superposição das funções de onda \(\frac{i}{\sqrt{2}}(\psi_{21-1} + \psi_{211})\):
\[ {\rm 2p_y} = \frac{1}{\sqrt{32\pi}} \left( \frac{Z}{a} \right)^{\frac{5}{2}} r e^{-Zr/2a} \sin{\theta} \sin{\phi} . \tag{1.4} \]
E, o orbital \({\rm 2p_z}\) é a própria função de onda \(\psi_{210}\), ele não é fruto de superposição:
\[ {\rm 2p_z} = \frac{1}{\sqrt{32\pi}} \left( \frac{Z}{a} \right)^{\frac{5}{2}} r e^{-Zr/2a} \cos{\theta} . \tag{1.5} \]
Ademais, as funções de onda \(\psi_{211}\) e \(\psi_{21-1}\) são chamadas de orbitais \({\rm 2p_{+1}}\) e \({\rm 2p_{-1}}\), respectivamente (Peter Atkins 2011).
No geral, a função de onda é escrita como \(\psi_{nlm}\), onde: \(n\) é o número quântico principal, \(l\) é o momento angular orbital, e \(m\) é a projeção do momento angular orbital no eixo \(z\). Assim, as funções de onda \(\psi_{211}\), \(\psi_{210}\) e \(\psi_{21-1}\) possuem \(n=2\), \(l=1\) e projeções \(m=1,0,-1\), respectivamente.
Do ponto de vista dos orbitais, os números \(l\) recebem nomes especiais, dados pela convenção de espectroscopia atômica:
\[ \begin{aligned} l &= 0 \implies s ,\\ l &= 1 \implies p ,\\ l &= 2 \implies d ,\\ l &= 3 \implies f ,\\ l &= 4 \implies g ,\\ l &= 5 \implies h . \end{aligned} \tag{1.6} \]
Por isso, os orbitais \({\rm s}\) são orbitais de momento angular orbital \(l=0\), os orbitais \({\rm p}\) são orbitais de momento angular orbital \(l=1\), e assim por diante.
Percebe-se que as equações (1.3), (1.4) e (1.5) são funções de três variáveis, pois a distância do elétron ao núcleo depende das coordenadas do elétron em relação ao núcleo:
\[ r = \sqrt{x^2 + y^2 + z^2} . \tag{1.7} \]
Os ângulos \(\theta\) e \(\phi\) também dependem das coordenadas do elétron em relação ao núcleo:
\[ \begin{aligned} \theta &= \arccos{ (z/r) } ,\\ \phi &= \arctan{ (y/x) } . \end{aligned} \tag{1.8} \]
O artigo foi escrito com o objetivo de compartilhar a técnica computacional envolvida na visualização de funções com 3 variáveis. Como todos sabem, para se graficar uma função com 3 variáveis necessitaríamos de um espaço com 4 eixos: 3 eixos para as variáveis \((x,y,z)\), e um quarto eixo para se colocar os valores da função \(f(x,y,z)\). Fica claro que não há possibilidade de graficar esse tipo de função da mesma maneira que graficamos uma função com 2 variáveis: utilizando 2 eixos para as variáveis \((x,y)\), e um terceiro eixo com o valor da função \(f(x,y)\).
No caso das funções com 3 variáveis, deve-se utilizar métodos de visualização volumétrica, os quais criam uma superfície no espaço, de pontos \((x,y,z)\), sendo que, ao percorrer essa superfície, a função \(f(x,y,z)\) é uma constante. Em outras palvras, contrói-se uma superfície tridimensional na qual \(f(x,y,z) = \alpha\), sendo \(\alpha\) um parâmetro. As superfícies de contorno tridimensionais assim construídas são chamadas isosuperfícies.
Vamos utilizar como método de visualização de isosuperfícies,
o algoritmo dos cubos marchando (marching cubes algorithm),
desenvolvido por Lorensen e Cline (W E Lorensen 1987).
Este algoritmo faz parte do utilitário contour3d, do pacote misc3d,
desenvolvido por Feng and Tierney para a linguagem de programação R (Dai Feng 2008).
2 Os elementos de trabalho
Antes de começarmos a fazer os gráficos dos orbitais, vamos entender os elementos de trabalho.
Estudaremos a estrutura eletrônica da molécula de hidrogênio, \({\rm H_2}\), formada por 2 núcleos de hidrogênio e por 2 elétrons. Os núcleos serão identificados por núcleo \({a}\) e núcleo \({b}\), e os elétrons por elétron \({1}\) e elétron \({2}\).
A Fig. 2.1 mostra a geometria do problema. Os núcleos foram colocados sobre o eixo \(z\) e estão separados pela distância \(R\). As distâncias dos elétrons \({i=1,2}\) até os núcleos \(a\) e \(b\) são representadas pelos vetores \({\mathbf{r}_{ai}}\) e \({\mathbf{r}_{bi}}\), de módulos:
\[ \begin{aligned} r _ {bi} &= \sqrt{x_i^2 + y_i^2 + (z_i - R)^2} ,\\ r _ {ai} &= \sqrt{x_i^2 + y_i^2 + z_i^2} . \end{aligned} \tag{2.1} \]
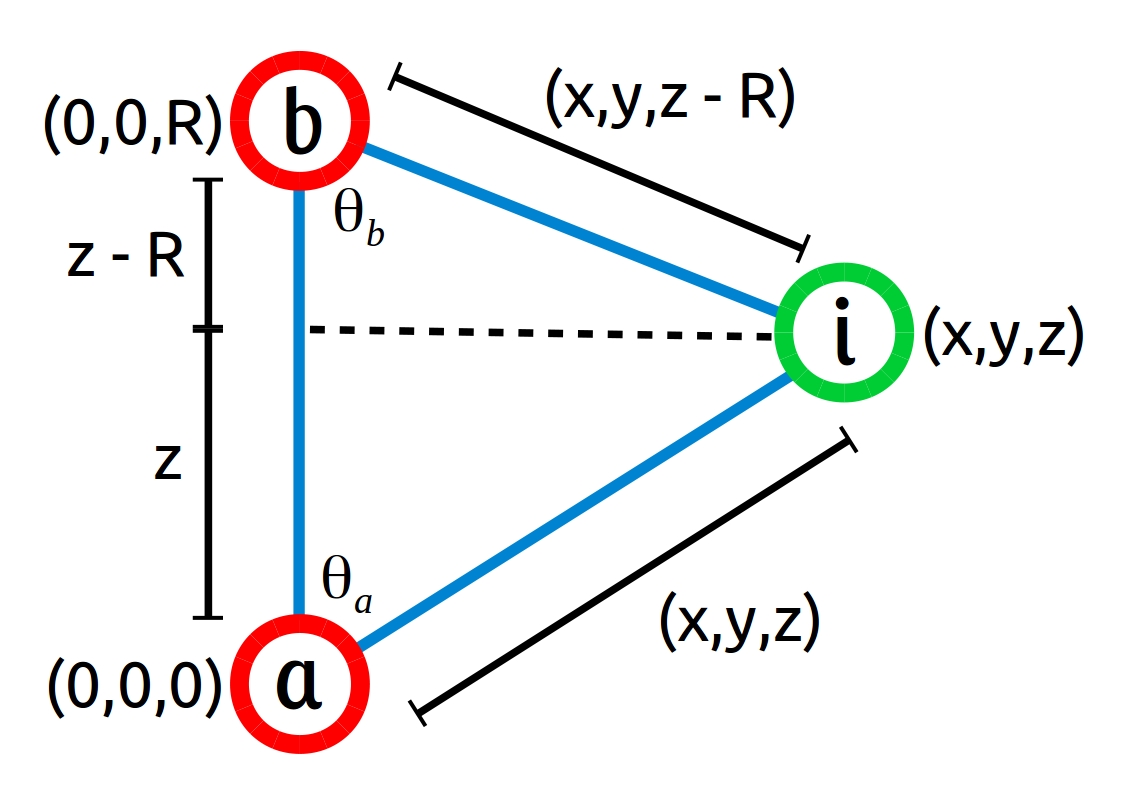
Figura 2.1: Distâncias e ângulos dos elétrons \({i=1,2}\) em relação aos núcleos \(a\) e \(b\).
Os ângulos que os elétrons \({i=1,2}\) fazem com o eixo \(z\), são:
\[ \begin{aligned} \theta _ {bi} &= \arccos{ [ (z_i - R) / r _ {bi} ] } ,\\ \theta _ {ai} &= \arccos{ [ z_i / r _ {ai} ] } . \end{aligned} \tag{2.2} \]
Os ângulos que os elétrons \({i=1,2}\) fazem com o eixo \(x\), são:
\[ \begin{aligned} \phi _ {bi} &= \arctan{ (y_i / x_i) } ,\\ \phi _ {ai} &= \arctan{ (y_i / x_i) } . \end{aligned} \tag{2.3} \]
3 A ocupação de orbitais
O elétron \({1}\) pode ocupar o orbital atômico do núcleo \({a}\). Vamos representar essa ocupação dizendo que \(\psi_a(1)\). Se o elétron \({1}\) está no orbital do núcleo \({a}\), é possível que o elétron \({2}\) esteja no orbital do núcleo \({b}\), então, é possível escrever \(\psi_b(2)\). Esta maneira de ocupar os orbitais dos átomos de hidrogênio proporciona que o orbital da molécula de \({\rm H_2}\) seja:
\[ \Psi = \psi_a(1) \psi_b(2) . \tag{3.1} \]
Todavia, também é possível que o elétron \({2}\) ocupe o orbital atômico do núcleo \({a}\) e o elétron \({1}\) ocupe o orbital do núcleo \({b}\), então, também é possível escrever \(\psi_a(2)\) e \(\psi_b(1)\), respectivamente. Esta nova configuração viabiliza que o \({\rm H_2}\) tenha o seguinte orbital molecular:
\[ \Psi = \psi_a(2) \psi_b(1) . \tag{3.2} \]
Visto que (3.1) e (3.2) são igualmente prováveis, o orbital molecular total do \({\rm H_2}\) deve ser a superposição das funções de onda de cada possibilidade (Peter Atkins 2011):
\[ \Psi = \psi_a(1) \psi_b(2) + \psi_a(2) \psi_b(1) . \tag{3.3} \]
Nota: A equação acima não está normalizada.
Vamos entender como utilizar a equação (3.3). A ideia é atribuir orbitais eletrônicos ao redor dos núcleos \(a\) e \(b\). Vamos supor que \(\psi_a = {\rm 2p_z}\) e que \(\psi_b = {\rm 2p_z}\). Substituindo a fórmula (1.5) em (3.3), e deixando de lado a constante \(\frac{1}{\sqrt{32\pi}} \left( \frac{1}{a} \right)^{\frac{5}{2}}\), tem-se:
\[ \begin{aligned} \Psi &= r _ {a1} e^{- r _ {a1} /2a} \cos{\theta _ {a1}} \times r _ {b2} e^{- r _ {b2} /2a} \cos{\theta _ {b2}} \\ &+ r _ {a2} e^{- r _ {a2} /2a} \cos{\theta _ {a2}} \times r _ {b1} e^{- r _ {b1} /2a} \cos{\theta _ {b1}} . \end{aligned} \tag{3.4} \]
Nota: Como estamos trabalhando com hidrogênios, \(Z = 1\).
As distâncias são determinadas pelas fórmulas (2.1):
\[ \begin{aligned} r _ {a1} &= \sqrt{x_1^2 + y_1^2 + z_1^2} ,\\ r _ {b2} &= \sqrt{x_2^2 + y_2^2 + (z_2 - R)^2} ,\\ r _ {a2} &= \sqrt{x_2^2 + y_2^2 + z_2^2} ,\\ r _ {b1} &= \sqrt{x_1^2 + y_1^2 + (z_1 - R)^2} . \end{aligned} \tag{3.5} \]
Já os ângulos são calculados pelas equações (2.2):
\[ \begin{aligned} \theta _ {a1} &= \arccos{ [ z_1 / r _ {a1} ] } ,\\ \theta _ {b2} &= \arccos{ [ (z_2 - R) / r _ {b2} ] } ,\\ \theta _ {a2} &= \arccos{ [ z_2 / r _ {a2} ] } ,\\ \theta _ {b1} &= \arccos{ [ (z_1 - R) / r _ {b1} ] } . \end{aligned} \tag{3.6} \]
4 O código fonte
Estamos com as ferramentas necessárias para dar início à escrita do código que vai gerar os gráficos das isosuperfícies.
A ideia central é fazer uma malha de pontos \((x,y,z)\)
com três comandos seq(-10,10,length=101), para alimentar as fórmulas dos orbitais.
Depois, escrever uma função com o comando function() que vai conter as fórmulas dos orbitais
e, em seguida, chamar o comando contour3d que se encarregará de fazer as isosuperfícies.
#Construção da malha espacial de pontos
x <- seq(-17,18,length=101)
y <- seq(-16,19,length=101)
z <- seq(-15,20,length=101)
#Função que contém as fórmulas dos orbitais
OrbMO <- function(x,y,z) {
x1 <- x
y1 <- y
z1 <- z
R <- 1.4
r_b1 <- sqrt(x1^2 + y1^2 + (z1-R)^2)
theta_b1 <- acos( (z1-R)/r_b1 )
phi_b1 <- atan2(y1,x1)
x2 <- x
y2 <- y
z2 <- z
r_b2 <- sqrt(x2^2 + y2^2 + (z2-R)^2)
theta_b2 <- acos( (z2-R)/r_b2 )
phi_b2 <- atan2(y2,x2)
r_a1 <- sqrt(x1^2 + y1^2 + z1^2)
theta_a1 <- acos(z1/r_a1)
phi_a1 <- atan2(y1, x1)
r_a2 <- sqrt(x2^2 + y2^2 + z2^2)
theta_a2 <- acos(z2/r_a2)
phi_a2 <- atan2(y2, x2)
b1 <- exp(-r_b1 ) /sqrt( pi) #1s
b2 <- exp(-r_b2 ) /sqrt( pi) #1s
a1 <- exp(-r_a1 ) /sqrt( pi) #1s
a2 <- exp(-r_a2 ) /sqrt( pi) #1s
b1 <- (2 - r_b1) * exp(-r_b1/2) /sqrt(32*pi) #2s
b2 <- (2 - r_b2) * exp(-r_b2/2) /sqrt(32*pi) #2s
a1 <- (2 - r_a1) * exp(-r_a1/2) /sqrt(32*pi) #2s
a2 <- (2 - r_a2) * exp(-r_a2/2) /sqrt(32*pi) #2s
b1 <- cos(theta_b1) * r_b1 * exp(-r_b1/2) /sqrt(32*pi) #2pz
b2 <- cos(theta_b2) * r_b2 * exp(-r_b2/2) /sqrt(32*pi) #2pz
a1 <- cos(theta_a1) * r_a1 * exp(-r_a1/2) /sqrt(32*pi) #2pz
a2 <- cos(theta_a2) * r_a2 * exp(-r_a2/2) /sqrt(32*pi) #2pz
b1 <- sin(theta_b1)*cos(phi_b1) * r_b1 * exp(-r_b1/2) /sqrt(32*pi) #2px
b2 <- sin(theta_b2)*cos(phi_b2) * r_b2 * exp(-r_b2/2) /sqrt(32*pi) #2px
a1 <- sin(theta_a1)*cos(phi_a1) * r_a1 * exp(-r_a1/2) /sqrt(32*pi) #2px
a2 <- sin(theta_a2)*cos(phi_a2) * r_a2 * exp(-r_a2/2) /sqrt(32*pi) #2px
a1 #Fórmula do orbital atômico
a1 * b2 + a2 * b1 #Fórmula do orbital molecular
}
contour3d(OrbMO, x,y,z, level=0.001) #Construção da isosuperfícieO programa é bem flexível,
pode-se comentar linhas para gerar isosuperfícies das mais variadas:
observe que há o bloco do orbital 1s, 2s, 2pz e 2px.
Lembrando que em R, os comentários são feitos com #.
Há duas opções de gráficos: (1) pode-se graficar o orbital atômico, deixando a linha a1 ativa;
ou (2) pode-se graficar o orbital molecular, deixando a linha a1*b2+a2*b1 ativa.
É o vamos fazer a partir de agora.
5 Os orbitais atômicos
Para graficar os orbitais atômicos, deve-se comentar a linha a1*b2+a2*b1.
Ademais, deve-se deixar ativo apenas o bloco do orbital que se deseja graficar.
Por exemplo, para graficar o orbital atômico \({\rm 1s}\),
deve-se comentar os blocos 2s, 2pz, 2px
(colocando # na frente de cada linha).
As figuras 5.1, 5.2, 5.3 e 5.4, apresentam os orbitais atômicos \({\rm 1s}\), \({\rm 2s}\), \({\rm 2p_z}\) e \({\rm 2p_x}\) do hidrogênio, respectivamente.
Por causa da fórmula do orbital \({\rm 2s}\) apresentar o termo \((2 - r)\), seu gráfico mostra duas cascas, enquanto que o gráfico do orbital \({\rm 1s}\) apresenta somente uma.
Na visualização dos orbitais \({\rm 2p_z}\) e \({\rm 2p_x}\),
foi utilizada a técnica de disparar várias isosuperfícies concentrícas.
Isso é feito atribuindo valores à opção level do comando contour3d.
Como o máximo e o mínimo desses orbitais ocorrem entre \((-0,\!07\) e \(0,\!07)\),
foi escolhido os seguintes valores:
cut <- c(-0.06, -0.04, -0.02, 0.02, 0.04, 0.06)
contour3d(OrbMO, x,y,z, level=cut)Nota: Confira no artigo (Dai Feng 2008)
mais exemplos de como utilizar o pacote misc3d e seu poderoso comando contour3d.
Fica claro que o orbital \({\rm 2p_z}\) aponta para o eixo \(z\), e o \({\rm 2p_x}\) para o eixo \(x\).
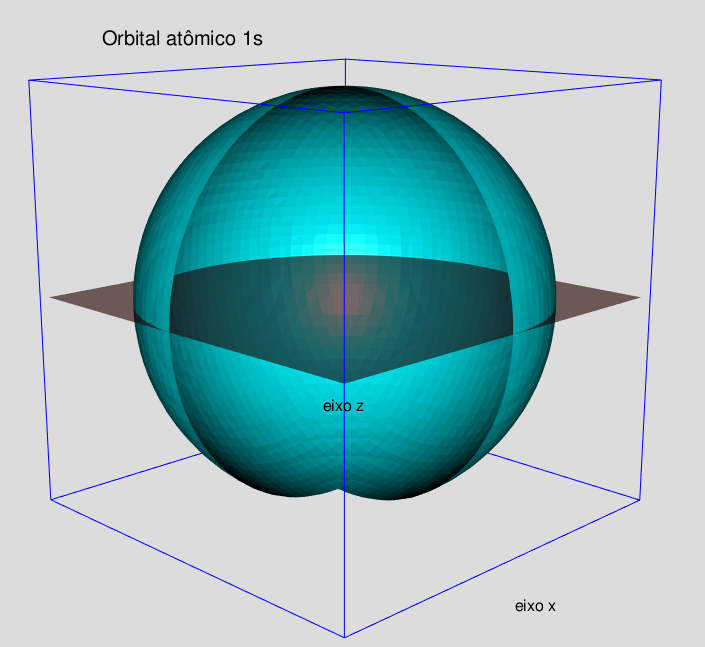
Figura 5.1: Núcleo de hidrogênio exibindo orbital \({\rm 1s}\).
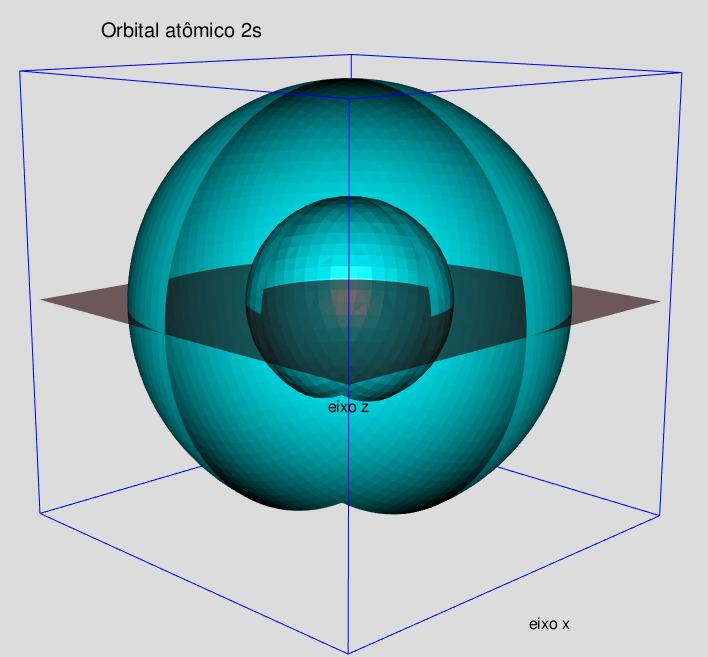
Figura 5.2: Núcleo de hidrogênio exibindo orbital \({\rm 2s}\).
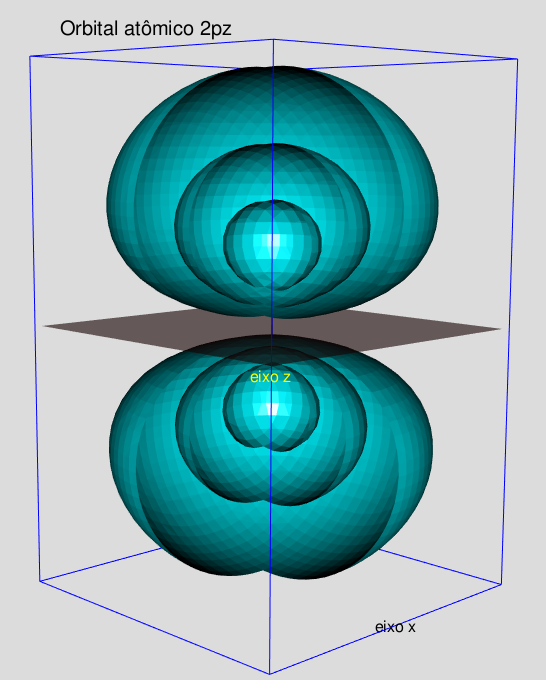
Figura 5.3: Núcleo de hidrogênio exibindo orbital \({\rm 2p_z}\): 3 isosuperfícies.
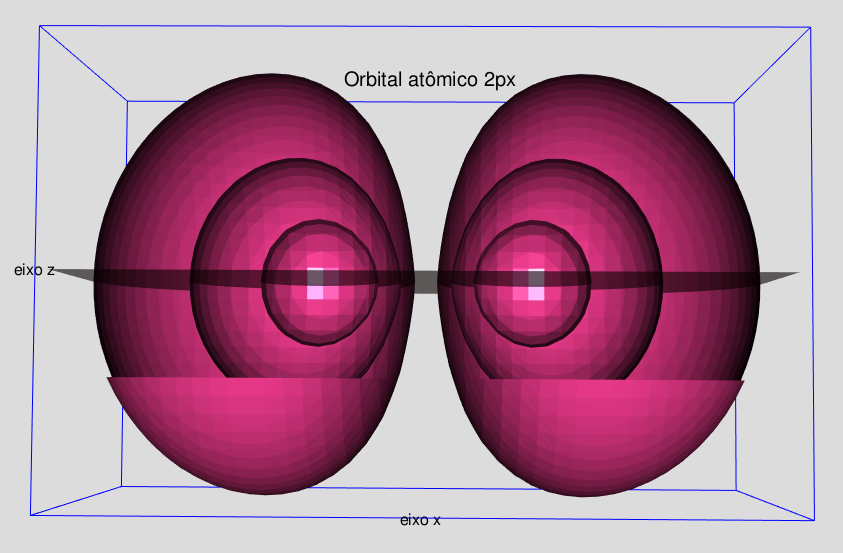
Figura 5.4: Núcleo de hidrogênio exibindo orbital \({\rm 2p_x}\): 3 isosuperfícies.
6 Os orbitais moleculares
Agora, para graficar os orbitais moleculares, deve-se comentar a linha a1.
Ademais, deve-se novamente deixar ativo apenas o bloco do orbital que se deseja graficar.
O valor experimental da distância entre os núcleos \(a\) e \(b\), da molécula de hidrogênio, é \(R = 1,\!40\ a_0\), segundo (Peter Atkins 2011).
Nota: Os orbitais estão sendo graficados ajustando \(a_0=1\) para efeito de simplificação.
As figuras 6.1, 6.2, 6.3 e 6.4, apresentam os orbitais moleculares da molécula de hidrogênio, com os núcleos \(a\) e \(b\) exibindo: ambos \({\rm 1s}\) \(({\rm 1s1s})\); ambos \({\rm 2s}\) \(({\rm 2s2s})\); ambos \({\rm 2p_z}\) \(({\rm 2p_z2p_z})\); e ambos \({\rm 2p_x}\) \(({\rm 2p_x2p_x})\).
Nota: Os casos \({\rm 2p_z2p_z}\) e \({\rm 2p_x2p_x}\) apresentam 3 isosuperfícies concentrícas.
Planos cortando a localização dos núcleos também foram graficados: para o núcleo \(a\), o plano é \((x,y,0)\), e para o núcleo \(b\), \((x,y,R)\). Os comandos para graficar os planos são:
planes3d(0,0,1, d=-1.4, col="red", alpha=0.6) #Plano sobre o núcleo "b"
planes3d(0,0,1, d= 0.0, col="red", alpha=0.6) #Plano sobre o núcleo "a"O orbital molecular do caso \({\rm 1s}\)\({\rm 1s}\) apresenta apenas 1 casca, enquanto que o do caso \({\rm 2s}\)\({\rm 2s}\), 3 cascas: decorrente da multiplicação de termos \((2 - r_a)\) e \((2 - r_b)\).
A ligação efetuada pelo orbital molecular \({\rm 2p_z}\)\({\rm 2p_z}\) é predominantemente na direção do eixo \(z\), enquando que a do \({\rm 2p_x}\)\({\rm 2p_x}\) é na direção do eixo \(x\).
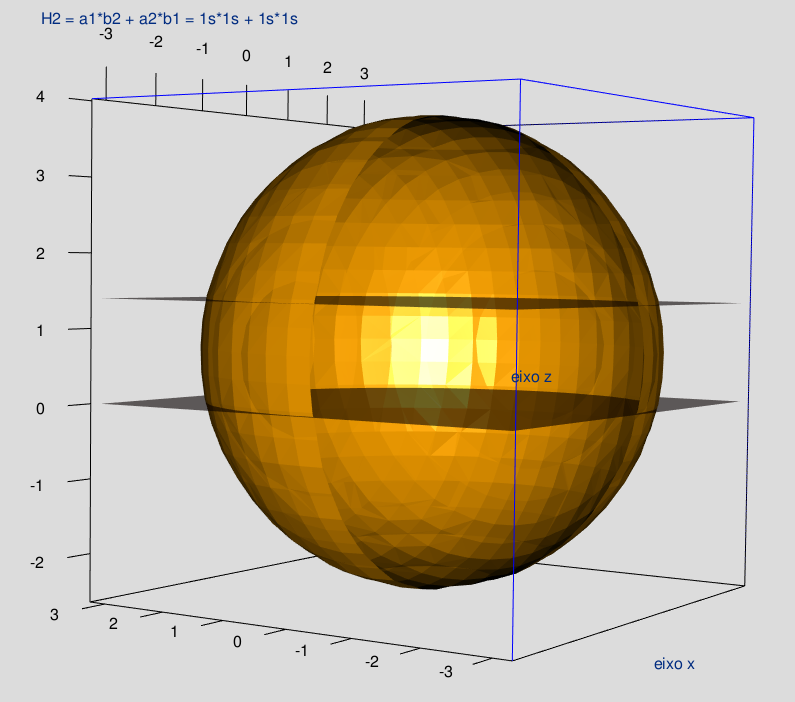
Figura 6.1: \({\rm H_2}\): Núcleos \(a\) e \(b\) na configuração \({\rm 1s1s}\).

Figura 6.2: \({\rm H_2}\): Núcleos \(a\) e \(b\) na configuração \({\rm 2s2s}\).
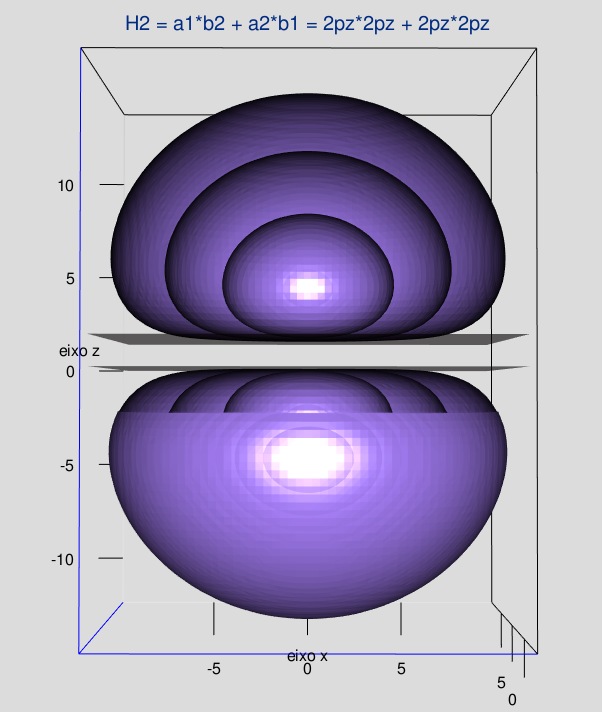
Figura 6.3: \({\rm H_2}\): Núcleos \(a\) e \(b\) na configuração \({\rm 2p_z2p_z}\): 3 isosuperfícies.
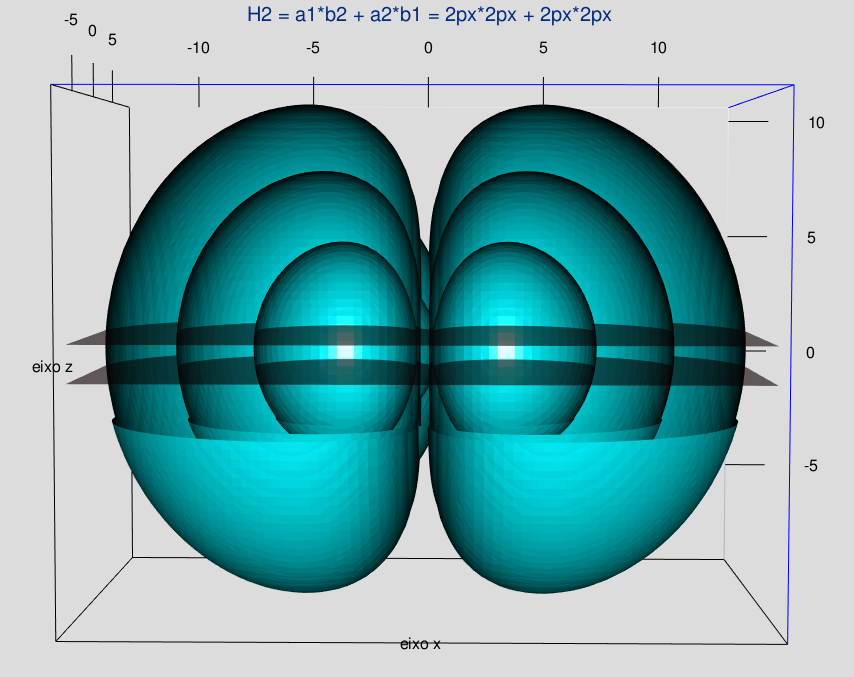
Figura 6.4: \({\rm H_2}\): Núcleos \(a\) e \(b\) na configuração \({\rm 2p_x2p_x}\): 3 isosuperfícies.
Vale ressaltar que, em um ambiente natural, é atípico a molécula de hidrogênio estar excitada a ponto de seus elétrons ocuparem o orbital molecular \({\rm 2p_z2p_z}\), ou \({\rm 2p_x2p_x}\). É comum que ela se encontre no estado fundamental: quando os 2 elétrons ocupam o orbital molecular \({\rm 1s1s}\). Já em um ambiente agitado, a molécula de hidrogênio pode ir para o primeiro estado excitado: construído com um elétron ocupando o orbital atômico \({\rm 1s}\), e o outro elétron, o \({\rm 2s}\), resultando na configuração molecular \({\rm 1s}\)\({\rm 2s}\). Este orbital misto também é possível de ser graficado acrescentando o seguinte bloco ao código fonte (ver figura 6.5):
b1 <- (2 - r_b1) * exp(-r_b1/2) /sqrt(32*pi) #2s
b2 <- (2 - r_b2) * exp(-r_b2/2) /sqrt(32*pi) #2s
a1 <- exp(-r_a1 ) /sqrt( pi) #1s
a2 <- exp(-r_a2 ) /sqrt( pi) #1s
Figura 6.5: Molécula de hidrogênio na configuração \({\rm 1s2s}\).
7 Conclusão
A visualisação de funções com 3 variáveis é um problema gráfico que exige técnicas computacionais avançadas.
Com a pesquisa do artigo (Dai Feng 2008),
pude notar que seria possível visualizar orbitais atômicos e moleculares utilizando a linguagem R.
O código fonte compartilhado é flexível e pode ser implemantado para outras situações.